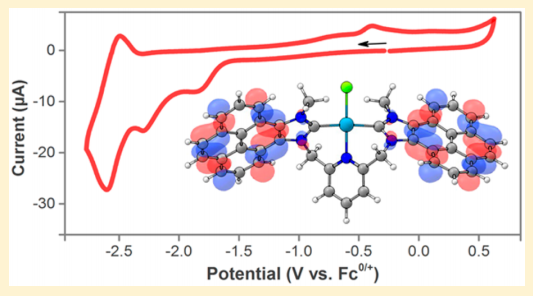
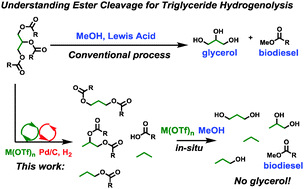
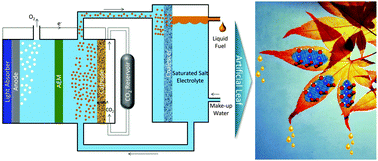
Light-Driven Heterogeneous Reduction of Carbon Dioxide:
Photocatalysts and Photoelectrodes
James L. White,† Maor F. Baruch,† James E. Pander III,† Yuan Hu,† Ivy C. Fortmeyer,† James Eujin Park,†
Tao Zhang,† Kuo Liao,† Jing Gu,‡ Yong Yan,‡ Travis W. Shaw,† Esta Abelev,† and Andrew B. Bocarsly*,†
†
Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States
‡
Chemical and Materials Science Center, National Renewable Energy Laboratory, Golden, Colorado 80401, United States
Chemical Reviews: http://pubs.acs.org/doi/pdf/10.1021/acs.chemrev.5b00370
DOI: 10.1021/acs.chemrev.5b00370
Abstract: Although modern photoelectrochemistry is often traced back to
1972 and the report by Honda and Fujishima1 that a TiO2
photoanode in an electrochemical cell caused the splitting of
water into O2 and H2 when illuminated, the first report of this
type of phenomenon dates back to Becquerel’s studies, published in 1839.2 This makes photoelectrochemistry one of
the oldest investigated techniques for the conversion of sunlight
into usable energy. Over this time frame, two general types of
photoelectrochemical cells have been developed. The first,
typified by Honda’s electrochemistry, is focused primarily on
the storage of light energy as high-energy chemical products.
Initially, this was termed “artificial photosynthesis” and was
focused for the most part on splitting water to generate H2 as
an environmentally benign fuel. The second type of photoelectrochemical
cell utilizes a chemically reversible redox couple
that undergoes a redox change of state at the photoelectrode,
followed by conversion of the product species back to the
reactant at the counter electrode. The net effect of this reaction
is a chemically invariant system that generates electricity from
light. The initial implementation of the Gratzel cell, which used ̈
a reversible I2/I3
− couple and a dye-sensitized TiO2 photoanode,
is an example of this type of system.3 The work under
consideration in this paper focuses on the photosynthetic cells
and related systems. However, an analysis of these systems, as is
more obviously critical to electricity-generating systems, must
take into account whether the system is merely catalytic for the
reaction of interest or is a system that actually converts light
energy into stored chemical energy. Thus, how one parametrizes
and evaluates a heterogeneous photoinduced charge
transfer process becomes a critical issue that is therefore
reviewed in this work.