Design of an artificial photosynthetic system for production of alcohols in high concentration from CO2
Meenesh R. Singha, Alexis T. Bell
Energy Environ. Sci., 2015, Advance Article
DOI: 10.1039/C5EE02783G
Received 10 Sep 2015, Accepted 06 Nov 2015
First published online 06 Nov 2015
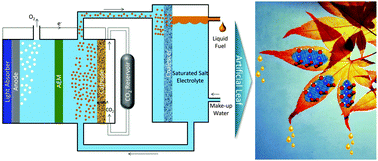
Abstract: Artificial photosynthesis of liquid fuels is a potential source for clean energy. Alcohols are particularly attractive products because of their high energy density and market value per amount of energy input. The major challenges in photo/electrochemical synthesis of alcohols from sunlight, water and CO2 are low product selectivity, high membrane fuel-crossover losses, and high cost of product separation from the electrolyte. Here we propose an artificial photosynthesis scheme for direct synthesis and separation to almost pure ethanol with minimum product crossover using saturated salt electrolytes. The ethanol produced in the saturated salt electrolytes can be readily phase separated into a microemulsion, which can be collected as pure products in a liquid–liquid extractor. A novel design of an integrated artificial photosynthetic system is proposed that continuously produces >90 wt% pure ethanol using a polycrystalline copper cathode at a current density of 0.85 mA cm−2. The annual production rate of >90 wt% ethanol using such a photosynthesis system operating at 10 mA cm−2 (12% solar-to-fuel (STF) efficiency) can be 15.27 million gallons per year per square kilometer, which corresponds to 7% of the industrial ethanol production capacity of California.