Remote site-selective C–H activation directed by a catalytic bifunctional template
Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USAZhipeng Zhang, Keita Tanaka & Jin-Quan Yu
Nature (2017) doi:10.1038/nature21418
http://www.nature.com/nature/journal/vaop/ncurrent/full/nature21418.html
Abstract
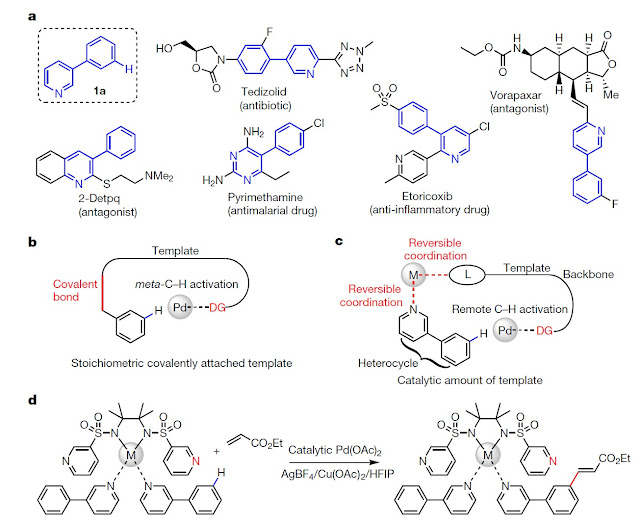
In chemical syntheses, the activation of carbon–hydrogen (C–H) bonds converts them directly into carbon–carbon or carbon–heteroatom bonds without requiring any prior functionalization. C–H activation can thus substantially reduce the number of steps involved in a synthesis. A single specific C–H bond in a substrate can be activated by using a ‘directing’ (usually a functional) group to obtain the desired product selectively1–5. The applicability of such a C–H activation reaction can be severely curtailed by the distance of the C–H bond in question from the directing group, and by the shape of the substrate, but several approaches have been developed to overcome these limitations6–12. In one such approach, an understanding of the distal and geometric relationships between the functional groups and C–H bonds of a substrate has been exploited to achieve meta-selective C–H activation by using a covalently attached, U-shaped template13–17. However, stoichiometric installation of this template has not been feasible in the absence of an appropriate functional group on which to attach it. Here we report the design of a catalytic, bifunctional nitrile template that binds a heterocyclic substrate via a reversible coordination instead of a covalent linkage. The two metal centres coordinated to this template have different roles: one reversibly anchors substrates near the catalyst, and the other cleaves remote C–H bonds. Using this strategy, we demonstrate remote, site-selective C–H olefination of heterocyclic substrates that do not have the necessary functional groups for covalently attaching templates.

