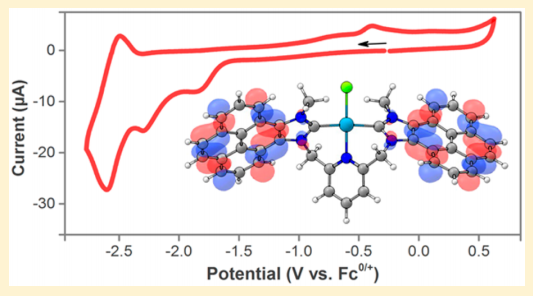
Standard Reduction Potentials for Oxygen and Carbon Dioxide
Couples in Acetonitrile and N,N‑Dimethylformamide
Michael L. Pegis,† John A. S. Roberts,‡,∥ Derek J. Wasylenko,§,⊥ Elizabeth A. Mader,† Aaron M. Appel,*,‡
and James M. Mayer*,†
†
Department of Chemistry, Yale University, PO Box 208107, New Haven, Connecticut 06520-8107, United States
‡
Center for Molecular Electrocatalysis, Pacific Northwest National Laboratory, P.O. Box 999 (K2-57), Richland, Washington 99352,
United States
§
Department of Chemistry, University of Washington, Campus Box 351700, Seattle, Washington 98195-1700, United States
Inorg. Chem. DOI: 10.1021/acs.inorgchem.5b02136
Abstract:
A variety of next-generation energy processes utilize the electrochemical interconversions of dioxygen and water
as the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER). Reported here are the first estimates of the
standard reduction potential of the O2 + 4e
− + 4H+ ⇋ 2H2O couple in organic solvents. The values are +1.21 V in acetonitrile
(MeCN) and +0.60 V in N,N-dimethylformamide (DMF), each versus the ferrocenium/ferrocene couple (Fc+/0) in the
respective solvent (as are all of the potentials reported here). The potentials have been determined using a thermochemical cycle
that combines the free energy for transferring water from aqueous solution to organic solvent, −0.43 kcal mol−1 for MeCN and
−1.47 kcal mol−1 for DMF, and the potential of the H+
/H2 couple, − 0.028 V in MeCN and −0.662 V in DMF. The H+
/H2
couple in DMF has been directly measured electrochemically using the previously reported procedure for the MeCN value. The
thermochemical approach used for the O2/H2O couple has been extended to the CO2/CO and CO2/CH4 couples to give values
of −0.12 and +0.15 V in MeCN and −0.73 and −0.48 V in DMF, respectively. Extensions to other reduction potentials are
discussed. Additionally, the free energy for transfer of protons from water to organic solvent is estimated as +14 kcal mol−1 for
acetonitrile and +0.6 kcal mol−1 for DMF.
TOC: